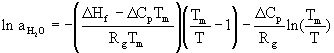 [1]
[1]
Kristin N. Duvall, James A. Dirksen and Terry A. Ring
Department of Chemical Engineering, University of Utah, 50 S Central Campus Drive, Salt Lake City, Utah 84112
To whom correspondence should be directed.
Introduction
Like any heat engine cycle, the Carnot efficiency (also called the "conversion factor" (1)) is given by difference in the high and low temperatures divided by the high temperature (1), h=(TH-TL/TH). The combustion source heats the LiBr solution between 200 ºC and 250 ºC (473 to 523 K). To improve the Carnot efficiency and to better cool the building low temperatures are desired. But since the concentrated LiBr solution can freeze in the heat exchanger, plugging it if cooled to too low a temperature, this limits the efficiency of the adsorption heat pump. The freezing point of concentrated LiBr solutions depends upon their concentration since different solids (e.g. ice, tri-, di- and mono- hydrates of LiBr) are produced. To over compensate for this problem the heat pump is operated in a way such its low temperature is several degrees above the equilibrium freezing point of the concentrated LiBr solution. However, when cooled a concentrated LiBr solution, like all pure salt solutions, does not freeze at the equilibrium freezing point but at a temperature below the equilibrium freezing point due to the need to supersaturate the solution before nucleation can take place. This difference in temperature is called the Ostwald-Meyers meta-stable zone (2): its size depends on the type of solution. Little is known about the parameters that control the meta-stable zone but it should depend upon surfaces present, cooling rate, ...
The equilibrium freezing point of LiBr solutions is given as a series of fit equations, see Table 1, for various LiBr× nH2O where n = 1, 2 and 3. This data is presented graphically in Figure 1 along with several experimentally measured equilibrium freezing points (3) found in the literature.
To establish the freezing point curve for ice, the water activity coefficient data by Patil et al. (4) were used and the equation (5)
[1]
where D Hf is the heat of fusion for water (+1,435.7 cal/mole), Rg is the gas constant, D Cp is the difference in heat capacity between ice and water (9.122 cal.mole-1.° C-1) at Tm, the normal freezing point of 273.15 K. The freezing point temperatures were calculated by inversion of the above formula for each water activity coefficient data point, which in turn was related to a particular solution concentration.
Figure 1 shows that the tri-hydrate, di-hydrate and mono-hydrate of lithium bromide have to be considered as the salt concentration increases. There are kinks in the solubility data which are indicative of a change in the solid phase crystallized. The liquid region is the region above the LiBr solubility curve and the equilibrium solubility of the ice curve. In addition, we also see a low temperature eutectic at a LiBr mass fraction of approximately 0.44 that freezes at » -25 ° C. This is the lowest temperature at which the LiBr solution should be liquid under equilibrium conditions. The solid crystallized at the eutectic is a mixture with about 56% ice and 44% LiBr3H2O using the lever rule.
The fit equations given in Table 1 can be reevaluated in terms of solubility products for each hydrated LiBr salt according to the equation:
![]()
![]() [2]
[2]
where ![]() are
the activity coefficients of the species in solution and
are
the activity coefficients of the species in solution and ![]() are
the mole fractions of species in solution. We can simplify this expression
by using the Gibbs-Duhem equation,
are
the mole fractions of species in solution. We can simplify this expression
by using the Gibbs-Duhem equation,
= 0 [3]
where Xi is the mole fraction of either
LiBr or H2O and gi
is the activity coefficient of LiBr (![]() )
or H2O (
)
or H2O (![]() ). Upon integration and
substitution of
). Upon integration and
substitution of ![]() , we find
, we find
![]() [4]
[4]
which can be then substituted into the solubility product, Ksp, giving
![]() [5]
[5]
This expression for Ksp is only a function of the mean ion activity coefficient and the solution mole fraction of LiBr. Using the solubility fit equations, the values of the various solubility products for the mono, di and tri-hydrates are given in Table 2 along with their uncertainties.
![]() [6]
[6]
where T40 is the initial bath temperature, R is the slope of the set point ramp and C[=hA0/m0Cp0] is a grouping of parameters consisting of the bath heat transfer characteristics, h is the convective heat transfer coefficient for the refrigeration coils in the bath, A0 is the bath heat transfer area, m0 is the thermal mass of the bath and Cp0 is the heat capacity for this thermal mass. The cooling inside the test tube immersed in this bath undergoing ramped cooling follows the theoretical equation:
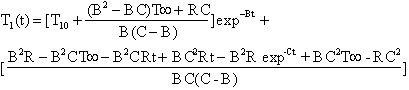 [7]
[7]
where T10 is the initial temperature of the fluid in the test tube, B [=U0A/mCp] is a group of parameters consisting of the test tube heat transfer characteristics, U0 is the overall heat transfer coefficient for heat transfer from the bath to the LiBr solution in the test tubes, A is the heat transfer area, m is the thermal mass of the glass vial and the LiBr solution and Cp is the heat capacity for the entire thermal mass. Since the LiBr solution was soaked at 50 ° C for 24 hrs both T10 and T40 are the same temperature for these experiments. A plot of the set point, bath temperature and test tube temperature is given in Figure 2. To determine the values of C and B best fits of the bath temperature and the test tube temperature without freezing are performed as a function of time using equations 6 and 7. Excellent fits with errors less than 0.1 ° C are obtained.
Freezing is observed due to the deviation of the temperature from the theoretical value since the heat of crystallization for LiBr salts is large and exothermic. According to theoretic work by Lesoult (9), e.g. equation 7, the onset of nucleation is observed for small deviations from the theoretical cooling curve. With this experimental system deviations of 0.4 ° C are easily detected, see Figure 3. As the nucleation rate increases and the crystal growth rate increases, larger amounts of the heat of crystallization are released and the temperature of the LiBr solution increases against the cooling ramp. After the nucleation rate and the crystal growth rate slow, the temperature then decreases as the cooling ramp in the bath overcomes the release of the heat of crystallization. The minimum in the temperature is easily observed. Deviations of 0.4 ° C from the theoretical curve equation 7 are obtained with a more detailed analysis. The 0.4 ° C deviation is typically only 0.5 ° C above the minimum temperature, this is easily detected due to the slope change that occurs at the minimum.
When undissolved crystals are present in solution due to insufficient solubilization another freezing point is measured. An example of the cooling curve for a solution with and without undissolved seed crystals is given in Figure 5. The freezing points for the solution with and without seed are easily distinguished. The 0.4 ° C deviation temperature for the solutions with crystals present from a previous freezing experiment reproduced (± 0.5 ° C) the equilibrium freezing point data from Freier (3) plotted in Figure 4.
The freezing point data at ramp rates of 20 ° C/hr and 0.2 ° C/hr were also measured. The results are shown in Figure 6 and compared with the equilibrium data for the crystallization of LiBr·2H2O and LiBr·3H2O. As the cooling rate was decreased, a higher freezing point was measured corresponding to a thinner Ostwald-Meyers instability region. This is expected as the equilibrium freezing point theoretically occurs for an infinitely slow cooling rate.
Crystallization induction times were measured by ramping down the temperature from 50 ° C to a given temperature above the observed freezing point at a 20 ° C/hr rate. This temperature was held for 24 hours waiting for an increase in the temperature to be observed indicating the freezing of the solution. The average induction times and their error bars for these experiments are plotted in Figure 7 as a function of the plateau temperature for a 60.54 wt % LiBr solution. As the temperature increases from the metastable freezing temperature of 3.42 ° C toward the equilibrium freezing point of 11.8 ° C for this solution, the induction time also increases. As the induction time increases, an increase of the corresponding standard deviation is observed.
In all cases, the crystals formed are large plate-like shaped. Crystals were observed to form first on the bottom glass surface of the test tube, suggesting surface nucleation, instead of homogeneous nucleation, to be the important nucleation step in these experiments. When the solution was completely frozen, the crystal filled 50 to 80% of the test tube depending upon the solution concentration.
![]() [8]
[8]
where D Hf is the heat of crystallization of the crystallizing solid, T is the actual temperature of crystallization, Teq is the equilibrium crystallization temperature and Rg is the gas constant. The heat of crystallization of the crystallizing solid at various solution concentrations was obtained from the solubility data using
![]() [9]
[9]
since the solubility fit equations are in a similar functional form, see Table 1. Using the experimentally observed freezing points, values of the critical supersaturation measured were typically, Sc » 1.1.
1) for homogeneous nucleation :
![]()
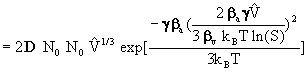 [10]
[10]
and 2) for surface nucleation :
![]() =
=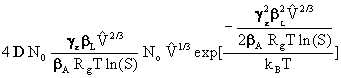 [11]
[11]
to determine a critical supersaturation that is responsible
for these humanly observable nucleation rates. In the above equations,
D
is
the solution diffusion coefficient, No is the number concentration
of LiBr in solution, r* is the critical radius for homogeneous
nucleation (subscript s is for surface nucleation), g
is
the surface energy for the crystal/solution interface, g?[»gd]
is the edge energy per unit length, ![]() is
the molar volume of the crystal. bv
and ba
are the volume and area conversion factors for homogeneous nucleation,
(for spherical nucleii, bv=4p
/3 and ba=4p
, for cubic nucleii, bv
=1 and ba=6),
bL
is the edge length conversion factor and bA
is the surface area conversion factor for surface nucleation (for a cylindrical
embryo of height, d/2, above the surface, bA=
p
and bL=2p
; for cubic embryo with an effective radius = L/2, bA=1
and bL=4),
kB is Boltzmann's constant.
is
the molar volume of the crystal. bv
and ba
are the volume and area conversion factors for homogeneous nucleation,
(for spherical nucleii, bv=4p
/3 and ba=4p
, for cubic nucleii, bv
=1 and ba=6),
bL
is the edge length conversion factor and bA
is the surface area conversion factor for surface nucleation (for a cylindrical
embryo of height, d/2, above the surface, bA=
p
and bL=2p
; for cubic embryo with an effective radius = L/2, bA=1
and bL=4),
kB is Boltzmann's constant.
The crystallization temperature was calculated by inverting equation
11 assuming that Is, the nucleation rate, equals 1 nucleus per cm2
per minute. This freezing temperature is calculated assuming surface nucleation
is responsible for spontaneous nucleation. The freezing temperature is
highly dependent upon the value of the surface energy ![]() used
for the foreign surface. A similar mathematical approach can be used for
the freezing temperature assuming homogeneous nucleation is responsible
for spontaneous nucleation.
used
for the foreign surface. A similar mathematical approach can be used for
the freezing temperature assuming homogeneous nucleation is responsible
for spontaneous nucleation.
There are two unknown values in these classical nucleation rate equations (equations 10-11): 1) the diffusion coefficient, D, however, it must have a value approximately 10-5 cm2/s for aqueous solutions, and 2) the surface energy of the crystal/solution interface, g. All other terms have standard values assuming a cubic nucleus. The nucleation rates are linear dependent on the diffusion coefficient, D; non-linear dependent on the surface energy, g, and on the temperature, especially in the definition of S in equation [8]. The effect of either errors in the value of D, used in calculating the crystallization temperature, or in the constant nucleation rate of 1 nucleus/cm2.min are smaller than the effect of errors in the crystallization temperature due to errors in the value of g. Thus the parameter g is the most critical parameter in determining the crystallization temperature using measurable nucleation rates.
The energy of the ice/water interface has been measured by Skapski et al. (10) to be 26 erg/cm2. (Note the data point for freezing ultra-pure water at 40 ° C (10) and fit with homogeneous nucleation curve in Figure 4.) Since the hydrated LiBr crystals will also have a layer of structured water at the surface, their surface energy is not likely to be very different from that of the ice/water interface. As a result this same value has been used for the surface energy of hydrated LiBr crystals undergoing homogeneous nucleation. The freezing point for an emulsion of concentrated LiBr solution in mineral oil measured by Dr. Austin Angel (12) is plotted in Figure 4 and is reasonably close to the homogeneous nucleation curve when one considers that errors in solution concentration can easily be made with this deliquescent salt. In an emulsion the solution droplets are more numerous than the dust particles present for heterogeneous nucleation to take place. For surface nucleation of hydrated LiBr salt at either the test tube surface or the thermowell surface, both made of pyrex glass, another interfacial energy should be used. This value was obtained from a best fit of the experimental freezing point data.
The experimental data points for our test tube experiments
support the hypothesis that surface nucleation on the test tube surface
is responsible for the nucleation event observed. For the surface nucleation
curve, data points for a solution with 60.54 wt % LiBr was used to determine
the appropriate value of the interfacial energy for surface nucleation.
This freezing point corresponds to 3.42 ±
0.56 ° C, where
the mean and standard deviation were determined with 69 separate freezing
point measurements for this solution at a ramp rate of 20 °
C/hr. Assuming Is to be one nucleus per cm2 per minute
the surface energy g was
solved to give a value of 40.0±1.2 erg/cm2. This value
was then used for all other surface nucleation curves, showing good agreement
with data for other concentrations where the di- and tri-hydrate crystallized
from solution. The standard deviation of the surface energy was calculated
by using the freezing point minus its standard deviation in equation [11]
and solving for the surface energy.
With the slower cooling rates the freezing point increases, moving closer to the equilibrium freezing point as would be expected since the equilibrium freezing point is obtained by an infinitely slow cooling rate. Classical nucleation theory, which was shown to successfully predict the meta-stable freezing point as a function of concentration in Figure 4, does not predict the effect of cooling rate. As a result, an important phenomenon is missing in classical nucleation theory - the effect of cooling rate. It is suggested that further work on nucleation theory be done to incorporate the effect of cooling rate so that the meta-stable region can be calculated for different cooling rates.
The induction times measured can be compared to the induction time for homogeneous nucleation given by (13)
![]() [12]
[12]
where n* is the number of molecules in the critical nucleus. This equation predicts that as S increases or the temperature is lowered below that of the freezing point, the value of the induction time decreases. A plot of this equation, not shown here, reveals the same shape, occurring at the same temperatures than the experimental data in Figure 7 but is four orders of magnitude lower. The reason for this discrepancy in crystallization induction times is probably due to limitations of classical nucleation theory, discussed above. The standard deviation of the crystallization induction time is also very large as shown in the error bars given in Figure 7. Since the induction time, t, depends upon the surface energy, g, we expect that heterogeneities in the surface energy lead to the large standard deviation in the induction time measured.
2. Freier, R. K., "Aqueous Solutions: data for inorganic and organic compounds", Vol. 1, 1976.
3. Elwell, D. and Scheel, H. J., "Crystal growth from high-temperature solutions, p.279, Academic Press, 1975.
4. Patil, K. R., Tripathi, A. D., Pathak, G. and Katti, S.S. , J. Chem. Eng. Data, 35 166-68 (1990).
5. Moelwyn-Hughes, E.A. ,"Physical Chemistry", 2nd edition, p. 836, Pergamon Press, New York, 1961.
6. Jang, S-M. and Myerson, A.S. in "Crystal Growth of Organic Materials" (A.S. Myerson, D.A. Green and P. Meenan, Eds.), ACS Conference Proceedings Series, p.53-58, American Chemical Society, Washington, D.C., 1996.
7. Zaltash, A., Ally, M.R., J. Chem. Eng. Data 37 110-113 (1992)
8. National Research Council, "International Critical Tables", (E.W. Washburn Ed.), Volume III, p.727, Mc Graw Hill, New York, 1928.
9. Lesoult, G., Castro, M. and Lacaze, J., Acta Mater. 46(3) 983-995 (1985).
10. Skapski, A., Billups, R. and Rooney, A., J. Chem. Phys., 26 1350 (1957).
11. Adamson, A.W., "Physical Chemistry of Surfaces" 5th edition, p.372, John Wiley & Sons, New York, 1990.
12. Private communication with Prof. Austin Angel, Dept. Materials Science, University of New Mexico, June 1996.
13. Becker, R. and Doring, W., Ann. Phys.(Leipzig), 24(5) 719-752 (1935)
14. Volmer, M. and Weber, A. , Z. Phys. Chem. 119 227 (1926)
15. Nielsen, A.E., "Kinetics of Precipitation", Pergamon Press, Oxford, (1964).
16. Adamson, A.W., "Physical Chemistry of Surfaces", 4th edition, p.322, John Wiley & Sons, New York, 1982.
17. Adamson, A.W., "Physical Chemistry of Surfaces", 4th edition, p.325, John Wiley & Sons, New York, 1982.
18. Wightman,. R.M. and Wipf, D.O., in "Electroanalytical Chemistry" (A.J. Bard, Ed.), Vol. 16, p. 267-353, Marcel Dekker, New York, 1988.
19. Dirksen, J.A. and Ring, T.A., Chemical Engineering Sci. 46[10] 2389-2427 (1991).
20. Broul, M., Nyyvlt, J and Sohnel, O., "Solubility in Inorganic Two-component Systems", Physical Sciences Data 6, Elsevier Scientific Publishing Co., Amsterdam, 1981.
21. Adamson, A.W., "Physical Chemistry of Surfaces", 5th edition, p.372, John Wiley & Sons, New York, 1990.
22. Austin, A. , Results taken from GRI annual report for 1996 on the project "Crystallization Inhibition in the LiBr-H2O system".
![]()
and the Molarity (in units of moles/Kg H2O), MLiBr, is given by :
![]()
where Mw is the molecular weight of either LiBr or H2O.
Classical Nucleation Theory
![]() [1]
[1]
![]()
![]() for cooling,
for cooling,
where v is the volume and a is the area of the aggregate,![]() is the molar volume of the precipitate, Rg is the gas constant, g
is the surface free energy per unit area and D
is the molar free energy for the phase transition [D
= -RgT ln(S) or with cooling of the vapor
is the molar volume of the precipitate, Rg is the gas constant, g
is the surface free energy per unit area and D
is the molar free energy for the phase transition [D
= -RgT ln(S) or with cooling of the vapor ![]() ]
where -D HT
is the enthalpy of the phase transition (either condensation for a gas
or fusion for a melt) occurring at temperature T below the equilibrium
temperature To. S [=a/ao] is the saturation ratio, where a is
the activity of the condensing species and ao is the activity in equilibrium
with the condensate. A generalized particle radius, r (=3v/a), can be used
to calculate the total free energy.
]
where -D HT
is the enthalpy of the phase transition (either condensation for a gas
or fusion for a melt) occurring at temperature T below the equilibrium
temperature To. S [=a/ao] is the saturation ratio, where a is
the activity of the condensing species and ao is the activity in equilibrium
with the condensate. A generalized particle radius, r (=3v/a), can be used
to calculate the total free energy.
D G(r) = -(bv r3/ )R T ln(S)+ gba r2, [2]
where bv is the volume conversion factor and ba is the surface area conversion factor. For a sphere, bv = 4p /3 and ba = 4p . For a cubic nuclei, bv = 8 and ba = 24. When the supersaturation, S<1.0, D G(r), is always positive and cluster formation is non spontaneous. When the supersaturation, S>1.0, D G(r) has a positive maximum at the critical size, r*, with a maximum Gibbs free energy, D Gmax = D G(r*). This is the energy barrier for nucleation. Clusters larger than the critical size will decrease their free energy by further growth, giving "stable nuclei" which grow to form macroscopic particles. Below the critical size, clusters will decrease their free energy by dissolving. The critical size, r*, is obtained by setting d /dr =0 giving
![]() [3]
[3]
Nielsen (15) has adopted the terminology that an
embryo is subcritical and a nucleus is supercritical in size. This critical
size corresponds to a value of the free energy at the maximum of![]() [4]
[4]
For a given value of S, all particles with r > r*
will grow and all particles with r< r* will disappear. Embryo ConcentrationsUsing
a Boltzmann distribution and these free-energy concepts, the equilibrium
number density of embryos of size, r, is given byNe(r) = No exp[![]() ]
=
]
= ![]() exp[
exp[![]() ]
[5]
]
[5]
where No is number of molecules per unit volume of condensing material in the gas phase and kB is Boltzmann's constant. This expression is only valid over a range of D G values where D G is greater than zero.Nucleation RateThe rate at which nuclei of critical size are assembled from an embryo and an additional atom is given by the reactionA(1 molecule) +B(r * - 1 molecule) ----> X(r *) [6]
which has the following rate assuming that only the
forward reaction is considered : ![]() [7]
[7]
where lAB
is the rate constant for the collision between molecules (A) of concentration,
No, with the units [![]() ] and nearly
critical clusters (B) of concentration, [
] and nearly
critical clusters (B) of concentration, [![]() ].
Equation [7] is a simple result of the classical nucleation theory for
the homogeneous nucleation rate with units of [
].
Equation [7] is a simple result of the classical nucleation theory for
the homogeneous nucleation rate with units of [![]() ].
The collision rate constant for two different gas molecules is available
from the kinetic theory of gases (16) :
].
The collision rate constant for two different gas molecules is available
from the kinetic theory of gases (16) :![]()
![]() [8]
[8]
where Mw is the molecular weight and s
is
the collision cross-section. The molecular weight correction term ![]() is
rarely used in Classical Nucleation theory. Becker and Doring (13) give
a more complicated expression for the collision rate as follows:lAB
= lAA(
is
rarely used in Classical Nucleation theory. Becker and Doring (13) give
a more complicated expression for the collision rate as follows:lAB
= lAA(![]() )1/2
[9]
)1/2
[9]
Often the Gibbs free energy for a cluster of size, r*- 1 molecule, and for a critical cluster of size , r* are assumed to be the same, i.e. D G(r*-1 molecule) »D G(r*) = D Gmax. As a result, the Classical Nucleation rate is given by :
![]() [10]
[10]
Classical Theory for MeltsTo alter Classical
Nucleation rate theory to be valid in either solution or melts, the collision
rate constant is altered in a way such as lAB
= 2 D d or lAB
= ![]() exp[
exp[![]() ]
[11]
]
[11]
where D is the diffusion coefficient, d is the center to center distance when two molecules collide, i.e. sAA =p /4 d2, for spheres and sAA = d2 for cubes, kB is Boltzmann's constant, h is Plank's constant and D GD is the Gibbs Free energy for diffusion in the solution or melt. Also note that for a melt of one component No a d-3.Finally, a useful concept is that of the critical supersaturation which is the value of S [=Sc] necessary to give a nucleation rate which is rapid on a basis humanly observable, say, 1 nuclei per cm3 per second (17).Surface Nucleation in a MeltWith surface nucleation, we have a collision between a diffusing molecule in the melt and a subcritical cluster attached to the surface. This collision rate will be different if the melt near the surface of the nuclei is stagnant or moving. If the melt is stagnant, molecular diffusion will transport the molecule to the surface. If the melt is moving, convective diffusion will transport the molecule to the surface. Let us assume that the surface is a disc of radius, Rd. When the melt is stagnant, the flux to a single subcritical surface cluster can be calculated by analogy to the current to an ultramicroelectrode (18) of radius, r :
j [ ] = 4 D No r [12]
where D is the diffusion coefficient. The radius of the ultramicroelectrode in the case of surface nucleation is analogous to the radius of the subcritical cluster attached to the surface of radius, rs* - 1 molecule. Calculating the flux to all these subcritical clusters at the surface, the surface nucleation rate can be approximated by :
Is [![]() ] =
4 D No rs* No d exp[
] =
4 D No rs* No d exp[![]() ]
= lsD
No No d exp[
]
= lsD
No No d exp[![]() ] [13]
] [13]
where the term No d exp[![]() ]
is the surface concentration [
]
is the surface concentration [![]() ]
of critical nuclei of size rs* and lsD
[=4 D rs*] is the collision rate constant for diffusion. Note that
the term Nod is the concentration of molecules at the surface. The above
surface nucleation rate is not that typically used for surface nucleation
but is faster than that given by the extension of classical nucleation
theory to surfaces (19) :
]
of critical nuclei of size rs* and lsD
[=4 D rs*] is the collision rate constant for diffusion. Note that
the term Nod is the concentration of molecules at the surface. The above
surface nucleation rate is not that typically used for surface nucleation
but is faster than that given by the extension of classical nucleation
theory to surfaces (19) :
Is [![]() ] = D d No
No d exp[
] = D d No
No d exp[![]() ] = lsCL
No No d exp[
] = lsCL
No No d exp[![]() ] [14]
] [14]
since each sub critical cluster has a highly curved diffusion profile and not a uniform diffusion profile as shown in Figures 1 and 2. At this juncture it is necessary to note that surface nucleation has a different definition of the Gibbs free energy from that for homogeneous nucleation,
D Gs [![]() ]
= -
]
= - ![]() RgT ln(S)+
g?bL
r [15]
RgT ln(S)+
g?bL
r [15]
where ge [»gd] is the edge energy per unit length. For a cylindrical embryo of height, d/2, off the surface, bA= p and bL=2p . For cubic embryo with an effective radius = L/2, bA=1 and bL=4. When the supersaturation, S<1.0, D Gs(r) is always positive and cluster formation is non-spontaneous. When the supersaturation, S>1.0, D Gs(r) has a positive maximum at the critical size, rs *, with a maximum Gibbs free energy, D Gs rmax = D G(rs *). This is again the energy barrier for nucleation.
TABLES
TABLE 1 Solubility Fit Equations (20) for LiBr.nH2O salts.
|
|
|
|
|
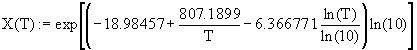 |
|
|
 |
|
|
 |
TABLE 2 Solubility Products for LiBrnH2O Salts.
These results assume that the mean ionic activity coefficient, g±,
is 1.0.
|
|
|
|
|
|
|
|
|
|
|
|
List of figures
Figure 1 Freezing Points of LiBr solutions or LiBr Solubility [Mass fraction LiBr] as a function of temperature. The solid precipitating out of solution is different depending upon the temperature range of interest. The dihydrate is precipitated from 5.7 ° C to 34.6 ° C at 1 atm. pressure, the monohydrate above 34.6 ° C and the trihydrate below 5.7 ° C. Ice freezing identified at low LiBr concentrations. Some ice freezing point data (+) have been calculated from solvent activity coefficient data. For the LiBrnH2O solubility, theory (lines) and experiment (?) agree nicely.
Figure 2 Plot of the set point, Ts=trace 1, bath temperature, Tb=trace 2, and test tube temperature, T1=trace 3 as a function of time.
Figure 3 Plot of experimental test tube temperatures when freezing is observed. T1b= bath temperature, T1m= measured test tube temperature, Tma, tma = maximum temperature and time, respectively; Tee, te = minimum temperature and time, respectively; Tc, tc = 0.4 ° C deviation temperature and time, respectively.
Figure 4 Equilibrium freezing point and crystallization temperatures versus weight fraction LiBr aqueous solution. Upper curves indicate equilibrium data for the crystallization of H2O(s), LiBr3H2O(s) and LiBr2H2O(s). Middle curves indicate critical temperature, Tsc, (° C) for the surface nucleation of crystals of H2O(s), LiBr3H2O(s) and LiBr2H2O(s) on glass surfaces. Bottom curves indicate critical temperature, Tc, (° C) for the homogeneous nucleation of crystals of H2O(s), LiBr3H2O(s) and LiBr2H2O(s). Experimental results for homogeneous nucleation of water gives 40 ° C (21) and 60 wt % LiBr solution (0.24 mole fraction) (in emulsion) gives 70 ° C (22) crystallization points. Other experimental data point come from present work. The average value of the freezing point is marked by the (+) symbol and the standard deviation is given by (?) symbols.
Figure 5 Cooling curve for a 61% LiBr solution with and without insoluble particles (of LiBr2H2O). In this case the temperature was lowered from 50 ° C to 30 ° C where it was held for 25 min. and then cooled at 20 ° C/hr to -10° C. This temperature plateau at 30 ° C was not used in subsequent experiments.
Figure 6 Equilibrium freezing point and crystallization temperatures versus weight fraction LiBr aqueous solution. Upper curves indicate equilibrium data for the crystallization of LiBr3H2O(s) and LiBr2H2O(s). Middle curve indicates the experimental freezing point due to cooling ramp at 0.2 ° C/hr and bottom curve indicates the experimental freezing point due to cooling ramp of 20 ° C/hr.
Figure 7 Induction time for 60.54 wt % LiBr solution at various temperatures
slighty above the measured freezing point (3.42 °
C).
Figures
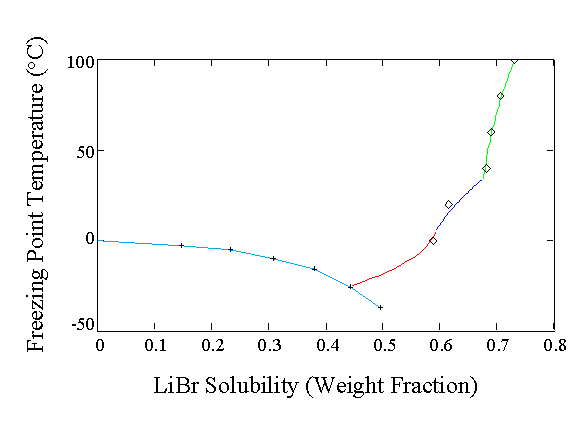
Figure 1 Freezing
Points of LiBr solutions or LiBr Solubility [Mass fraction LiBr] as a function
of temperature. The solid precipitating out of solution is different depending
upon the temperature range of interest. The dihydrate is precipitated from
5.7 ° C to 34.6 °
C at 1 atm. pressure, the monohydrate above 34.6 °
C and the trihydrate below 5.7 ° C. Ice
freezing identified at low LiBr concentrations. Some ice freezing point
data (+) have been calculated from solvent activity coefficient data. For
the LiBrnH2O solubility, theory (lines) and experiment (à
) agree nicely.

Figure 2 Plot of the
set point, Ts, bath temperature, Tb, and test tube
temperature, T1 as a function of time.
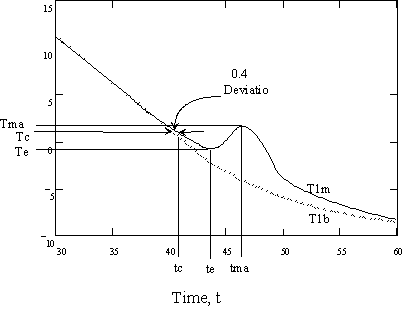 F
F
igure 3 Plot of experimental
test tube temperatures when freezing is observed. T1b= bath temperature,
T1m= measured test tube temperature, Tma, tma = maximum temperature and
time, respectively; Tee, te = minimum temperature and time, respectively;
Tc, tc = 0.4 ° C deviation temperature and
time, respectively.

Figure 4 Equilibrium
freezing point and crystallization temperatures versus weight fraction
LiBr aqueous solution. Upper curves indicate equilibrium data for the crystallization
of H2O(s), LiBr3H2O(s) and LiBr2H2O(s). Middle
curves indicate critical temperature, Tsc (°
C) for the surface nucleation of crystals of H2O(s), LiBr3H2O(s)
and LiBr2H2O(s) on glass surfaces. Bottom curves indicate critical
temperature, Tc, (° C) for the homogeneous
nucleation of crystals of H2O(s), LiBr3H2O(s) and
LiBr2H2O(s). Experimental results for homogeneous nucleation
of water gives 40 ° C (21) and 60 wt %
LiBr solution (0.24 mole fraction) (in emulsion) gives 70 °
C (22) crystallization points. Other experimental data point come from
present work. The average value of the freezing point is marked by the
(+) symbol and the standard deviation is given by (à
) symbols.

Figure 5 Cooling curve
for a 61% LiBr solution with and without insoluble particles (of LiBr2H2O).
In this case the temperature was lowered from 50 °
C to 30 ° C where it was held for 25 min.
and then cooled at 20 ° C/hr to -10°
C. This temperature plateau at 30 ° C was
not used in subsequent experiments.
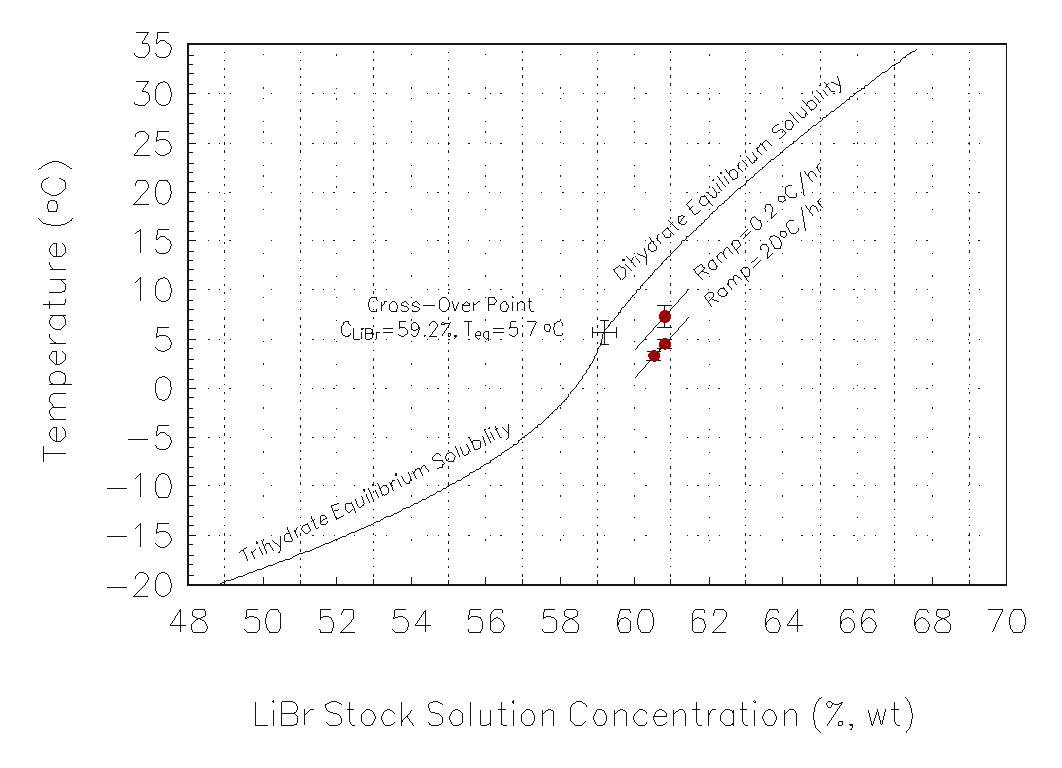
Figure 6 Equilibrium
freezing point and crystallization temperatures versus weight fraction
LiBr aqueous solution. Upper curves indicate equilibrium data for the crystallization
of LiBr3H2O(s) and LiBr2H2O(s). Middle curve indicates
the experimental freezing point due to cooling ramp at 0.2 °
C/hr and bottom curve indicates the experimental freezing point due to
cooling ramp of 20 ° C/hr.
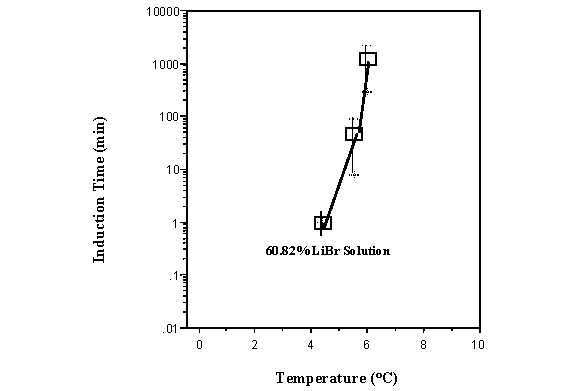
Figure 7 Induction
time for 60.54 wt % LiBr solution at various temperatures slighty above
the measured freezing point (3.42 ° C).